Gene Selection
毕业典礼还是完美的睡过去了(叹气,毕业之后成为又丧又废的NEET。
这半年主要都在折腾毕设,课题还是十分有趣的说。虽然被自己搞得杂乱不堪
有种自己的大学变成了37,990,696 字节的感觉。
十分糟糕的心情,毕业开始丧。听着更加丧的歌总结一下毕设。
十分传统的绪论
微阵列技术是用于分析基因表达的工具,可用于测定单个样品中的基因表达,或比较两种不同细胞类型或组织样品(如健康和癌组织)中的基因表达,近年来被广泛用于肿瘤细胞分子水平上的分类和预测研究。
在传统医疗中,很难只根据病理检测结果来下一个准确的诊断。而微阵列技术能够有效的对肿瘤进行识别和诊断。微阵列数据包含了数量庞大的基因,和非常少量的样本。基因表达谱中包含许多的噪声基因和冗余基因,因此需要通过特征选择的方法对数据进行筛选,这一识别过程即为基因选择。
特征选择是采用某种优化算法从微阵列数据中的所有属性特征里选出最具识别能力的基因子集的过程。特征选择有很多优点:
减小尺寸以降低计算成本
降低噪音,提高分类精度;
能取得更多可解释的特征,来帮助识别和监测目标疾病或功能类型。
因此各种聚类,分类和预测技术已经被用来分析,分类和了解基因表达数据,如 Fisher 判别分析 ,人工神经网络 ,以及支持向量机等。通过基于表达谱分类识别算法,再利用分类器构建预测分类模型。
因此设计本系统的目的在于提供一个可视化界面供研究人员直观的操控海量基因数据,选择不同的特征选择算法,直接获取结果。
图与脑洞
实在过于懒惰UI直接用R实现赞美bootstrap,然后拖延症导致很多脑洞没实现,导师强烈要求(划掉)加上用户管理以及存储功能,其实本来只打算完成一个简单的基因选择的功能和结果可视化对比。
继续懒惰的一个一个模块堆好了界面。
无意义其实可以跳过
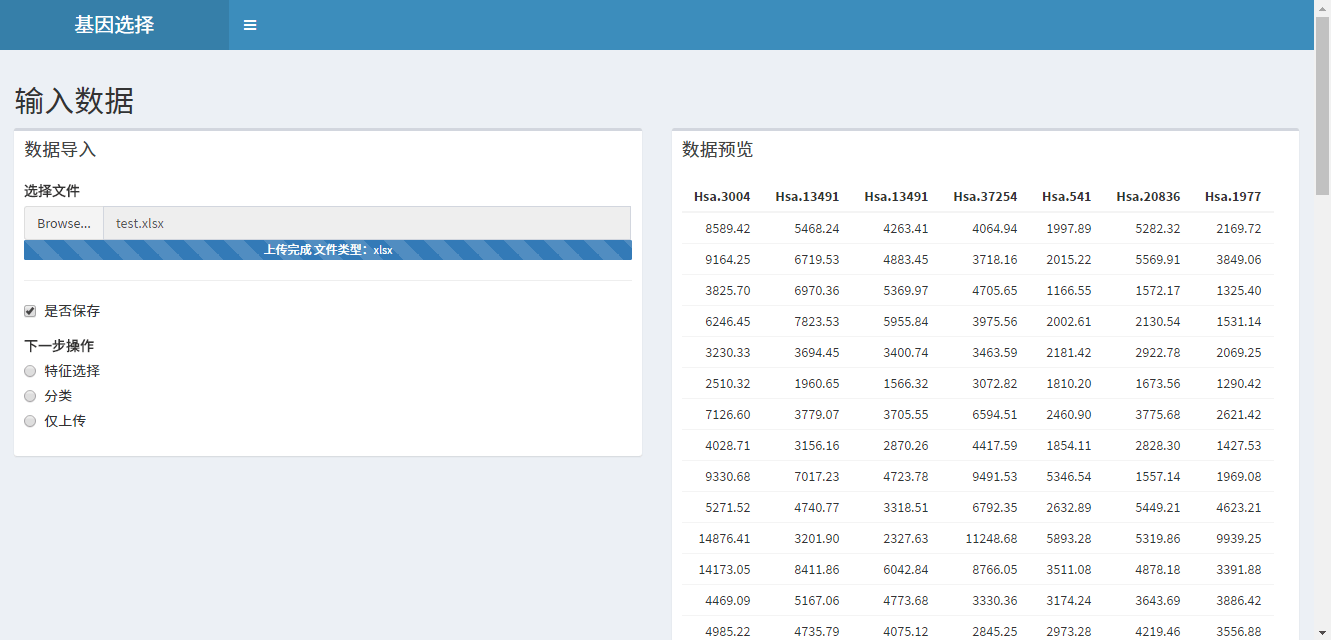
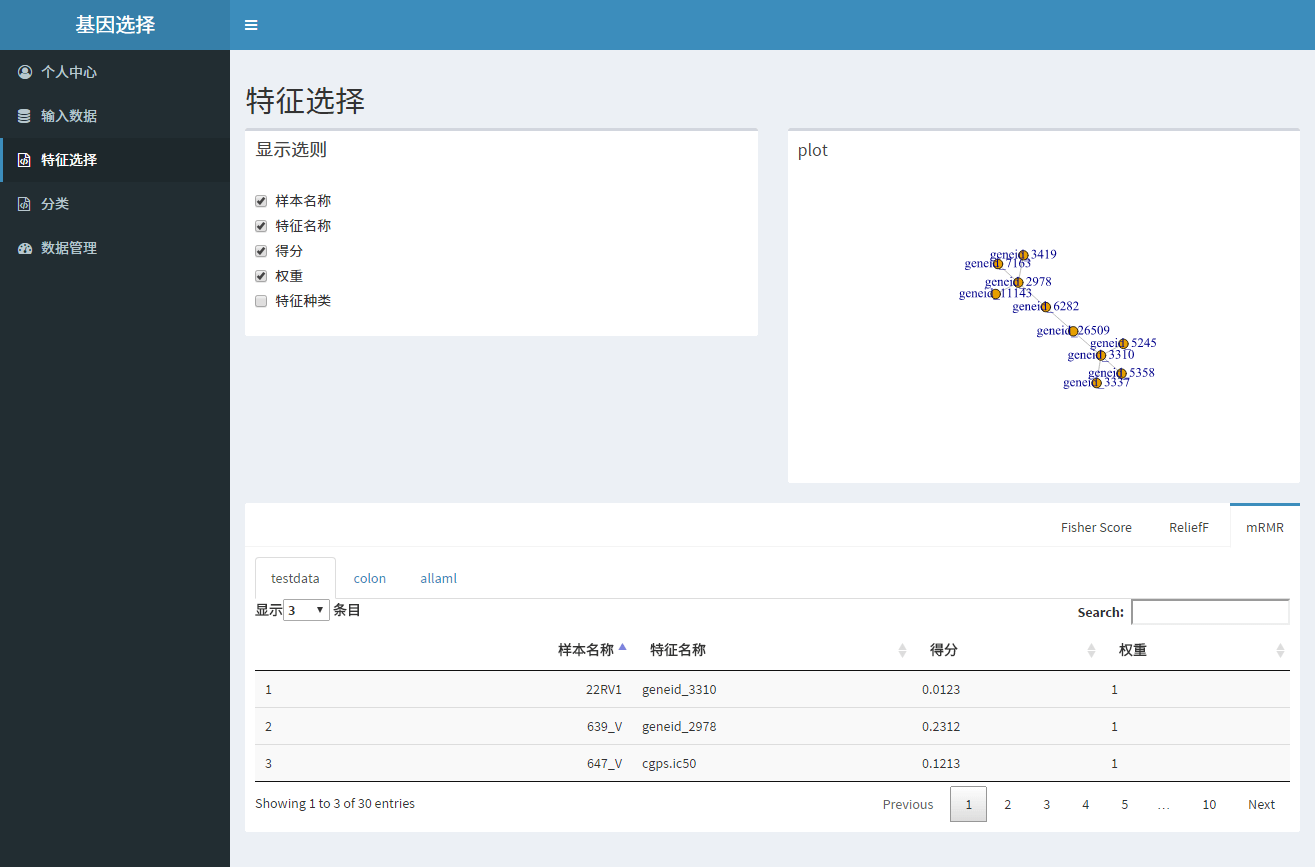
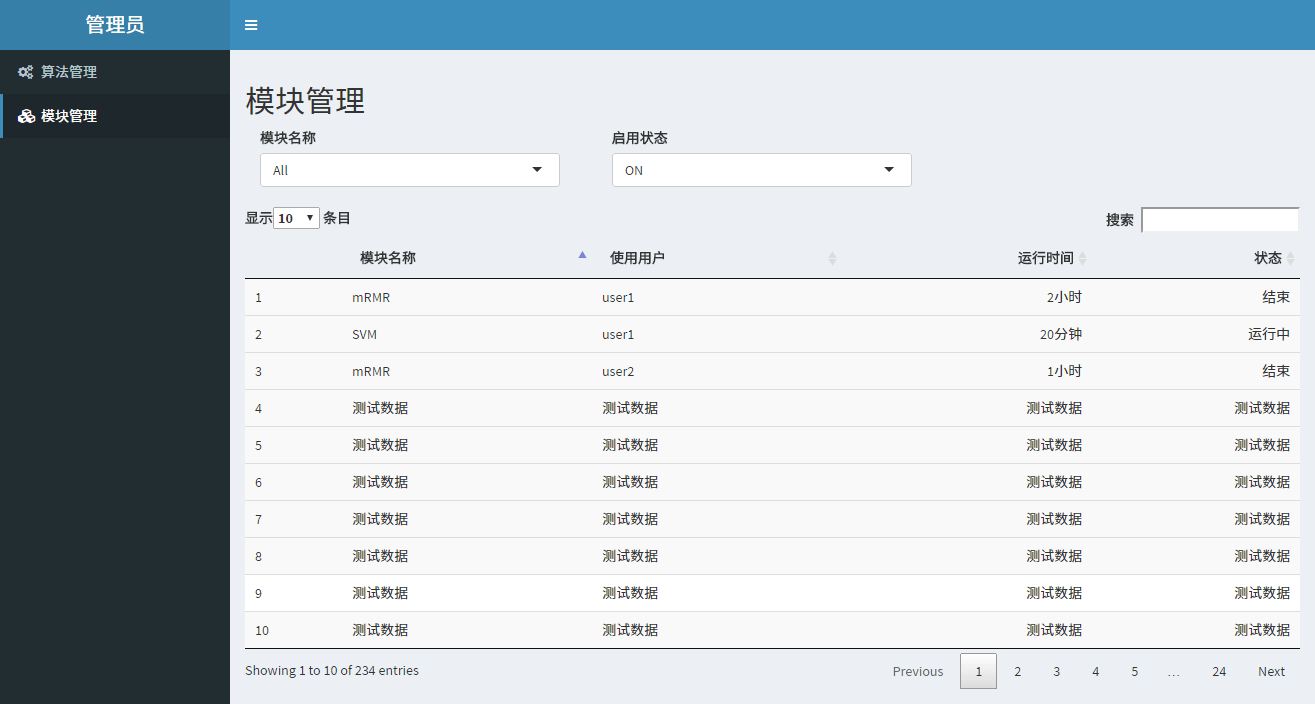
有点意义的笔记
算法概述
mRMR
令\(S\)表示选定基因的子集,\(\Omega\)是所有可用基因的集合;最小冗余可以通过计算得到\(\min _{S\subset \Omega }\dfrac {1}{\left|S\right| ^{2}}\sum_{i,j\in S}I(g_i, g_j) \)
其中\(I(g_i, g_j)\)是 \(i\)个 和 \(j\)个 基因之间的互信息即测量这两个相互依赖的变量。两个离散随机变量\(g_i\)和\(g_j\)的互信息可以被定义为\(\sum_{m\in g_i}\sum_{n\in g_j}p(m,n )log\dfrac{p(m,n)}{p(m)p(n)}\)
其中\(p(m, n)\)是\(g_i\)和\(g_j\)的联合概率分布函数,\(p(m)\)和\(p(n)\)分别是\(g_i\)和\(g_j\)的边际概率分布函数。 \(|S|\) 是\(S\)中基因数量,通常使用互信息\(I(T, g_j)\)来计算从基因$ g_i\(到类\) T = {t1,t2} \(的区分能力,其中\)t_1\(和\)t_2\(表示健康和肿瘤类别。因此,最大相关性可以通过以下公式计算\){S}{i,jS}I(T, g_i)$
mRMR 特征选择标准可以得到如下差异形式:\(\max_{S\subset \Omega }\{\sum_{i\in S}I(T, g_i)-[\dfrac{1}{\left|S\right|}\sum_{i,j\in S}I(g_i, g_j)]\}\)
ReliefF
设训练数据集为 \(D\) ,样本抽取次数为 \(m\) ,特征权重的阈值 \(\delta\) ,最近邻样本个数 \(k\) ,输出为各个特性的特征权重 \(T\) 。 设所有特征权重为 0 , \(T\) 为空集。从 \(D\) 中随机选择一个样本 \(R\) ;再从 \(R\) 的同类样本集中找到 \(R\) 的 \(k\) 个最近邻 \(H_j(j=1,2,...,k)\) , 再从每一个不同类的样本集中找出 \(k\) 个 最近邻 \(M_j(C)\) 。循环 \(m\) 次后开始从 \(A=1\) 到 \(N\) 计算特征\(W(A)=W(A)-\sum^{k}_{j=1}diff(A,R,H_j)/(mk)+\sum_{C \in class(R)}[\dfrac{p(C)}{1-p(Class(R))}\sum^k_{j=1}diff(A,R,M_j(C))]/(mk)\)
Fisher Score
给定 \(m\) 个特征, 将输入的矩阵 \(\mathbf{X} \in \mathbb R^{d \times n}\) 缩小到 \(\mathbf{Z} \in \mathbb R^{m \times n}\) ,则 Fisher Score 如下计算\(F( \mathbf{Z})=tr\{(\mathbf{\widetilde{S}}_b)\}(\mathbf{\widetilde{S}}_t + \gamma \mathbf{I})^{-1}\)
其中 \(\gamma\) 是一个正规参数, \(\widetilde{S}_b\) 和 \(\widetilde{S}_t\) 如以下给出 \[ \widetilde{S}_b=\sum^c_{k=1}(\widetilde{\mu}_k-\widetilde{\mu})(\widetilde{\mu}_k-\widetilde{\mu})^T \] \[ \widetilde{S}_t=\sum^n_{i=1}(z_i-\widetilde{\mu})^T \]
不存在的前端
Shiny is a new package from RStudio that makes it incredibly easy to build interactive web applications with R.
可以说是非常便利了。只需要看官方的tutorial,再参考一下gallery里的范例,从此不用切图所以说前端就是伪需求。此外shinydashboard 、shinythemes 、shinyjs
、htmlwidgets能够继续扩展。
不存在的服务端
三种基因选择方法分别调用了mRMRe、CORElearn、MASS三个package实现。然而,其实没什么好写的不如直接看官方pdf。
越写越不想写就这样吧,反正我也不知道我写了什么东西